
Materials theory is an exciting and fast-paced interdisciplinary field involving the theoretical prediction of the structure and properties of materials to discover new materials with properties suitable for applications in various areas of science and engineering. Materials theory combines elements of materials science, physics, chemistry, and computer science and has applications in chemical, electrical, mechanical, and civil engineering.
With the move to smaller and smaller structures, theoretical and computational predictions of materials properties on the nanoscale can accelerate materials development and are crucial for progress in the areas nanoscience and nanotechnology.
Two-dimensional and nanostructured materials for electronic and energy applications
Two-dimensional (2D) materials, nanocrystals, and other nanomaterials present an area of enormous scientific growth – more than 30,000 research papers appear every year on these topics. The synthesis of novel nanomaterials and their assembly into mesoscale structures provide a path to the creation and design of materials with tunable electronic, physical, and chemical properties. By virtue of their tunable properties, nanomaterials are excellent candidates for use in a range of emerging nanotechnologies and offer opportunities for the design of completely new types of materials for applications in fields such as photovoltaics, lighting, catalysis, and electronics.
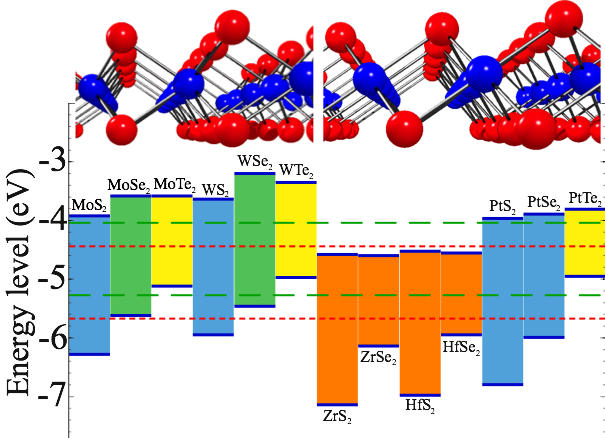
The last two decades have seen an explosion of interest in two-dimensional (2D) materials, which now can be synthesized in either single, or a few atomic layer form. Single layer materials not only represent the ultimate scaling in the vertical direction, but also show a variety of novel and useful electronic, optical, and mechanical properties. Through a data-mining based first-principles design approach we recently discovered a previously unrecognized family of single-layer materials of III-V materials and oxides and identified several chalcogenide compounds that are suitable for photocatalytic water splitting.
For nanocrystals, our research focuses on the computational prediction of the morphology of nanocrystals, their assembly into mesoscale structures, and their chemical transformation. For ligand-stabilized PbSe nanoparticles we discovered that variable ligand-surface coverage of nanocrystals controls their shape and their assembly into superlattice structures. For Co-P and Ni-P we predict that a change in diffusion mechanism leads to formation of hollow nanoparticles. For the organic semiconductor pentacene, we discover that a defect, called OH leads to long-lived electronic trap states that can degrade device performance and are sensitive to light.
![]()
Selected publications:
- Computational Discovery, Characterization, and Design of Single-Layer Materials.
H. L. Zhuang and R. G. Hennig. Journal of Metals 66, 366 (2014).
doi:10.1007/s11837-014-0885-3 - Single-Layer Group-III Monochalcogenide Photocatalysts for Water Splitting.
H. L. Zhuang and R. G. Hennig. Chem. Mater. 25, 3232 (2013).
doi:10.1021/cm401661x - Computational Discovery of Single-Layer III-V Materials.
H. L. Zhuang, A. K. Singh, and R. G. Hennig. Phys. Rev. B 87, 165415 (2013).
doi:10.1103/PhysRevB.87.165415 - Predicting Nanocrystal Shape Through Consideration of Surface-Ligand Interactions.
C. R. Bealing, W. J. Baumgardner, J. J. Choi, T. Hanrath, and R. G Hennig. ACS Nano 6, 2118 (2012).
doi:10.1021/nn3000466 - The structural evolution and diffusion during the chemical transformation from cobalt to cobalt phosphide nanoparticles.
D.-H. Ha, L. M. Moreau, C. R. Bealing, H. Zhang, R. G. Hennig, and R. D. Robinson. J. Mater. Chem. 21, 11498 (2011).
doi:C1JM10337G
Computational design of novel photocathodes
Part of the NSF funded Center for Bright Beams (CBB) we develop a high-throughput approach to identify materials and surfaces for photocathode applications that can potentially increase the brightness of photocathodes by orders of magnitude. Our contributions will be on the identification of novel photocathode materials and the optimization of compound superconductors for the RF accelerators. The photocathode materials will be screened for optimal electronic structure and quantum efficiency using density-functional theory. For the compound semiconductors we will determine thermodynamic and kinetic parameters to aid in the optimization of the microstructure. This work takes advantage of our MPInterfaces Python framework.
Computational methods for materials discovery and design
We systematically combine methods to both increase the accuracy and efficiently scan phase space to discover novel crystal structures, follow the evolution of defects and phases and predcit the assembly of hybrid materials. Our approaches rely on genetic algorithms to efficiently scan the phase space and discover new structures and transition pathways. We are developing the GASP code for structure prediction of materials. In addition we optimize fast empirical potential to extend te accuracy of electronic structure methods to larger length and time scales. Electronic structure methods such as density-functional theory estimates formation energies, diffusion and transition rates and describes electronic, optical, and mechanical properties of materials. Highly accurate quantum Monte-Carlo calculations benchmark the accuracy of approximations in electronic structure techniques. To describe systems in contact with solvents, we develop a solvation module VASPsol for the plane-wave density-functional code VASP
Selected publications:
- A grand canonical genetic algorithm for the prediction of multi-component phase diagrams and testing of empirical potentials.
W. W. Tipton and R. G. Hennig.
J. Phys.: Cond. Matter 25, 495401 (2013).
[web] - Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways.
K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias, and R. G. Hennig, submitted to J. Chem. Phys. (2013).
http://arxiv.org/abs/1310.4242. - A framework for solvation in quantum Monte Carlo.
K. A. Schwarz, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias, and R. G. Hennig.
Phys. Rev. B 85, 201102(R) (2012), Selected as Editor’s Suggestion.
[web] - Prediction of Structures, Phase Stability and Electrical Potential of Li-Si Battery Anode Materials.
W. W. Tipton, C. R. Bealing, K. Mathew, R. G. Hennig.
Phys. Rev. B 87, 184114 (2013).
[web]